AI-Enhanced Ultra-High Throughput Virtual Screening and Precision Docking
a. PlatformPlatform Overview
AlanDockAI™ is a next-generation molecular discovery platform that fuses a proprietary ultra-high-performance docking engine with quantum-mechanics–derived descriptors to deliver billion-scale virtual screening with unprecedented speed and precision. The platform dramatically outperforms conventional docking solutions, delivering orders-of-magnitude faster virtual screening while maintaining quantum-level accuracy in binding mode prediction. In parallel, Alan-Genion Quantum Tech has developed a suite of quantum mechanics (QM)–based structural elucidation tools, with a particular focus on computational spectroscopy for precise drug structure determination. By efficiently narrowing vast chemical spaces to high-confidence, biologically relevant hit compounds, AlanDockAI™ elevates virtual screening from a computational bottleneck to a strategic accelerator for drug discovery and molecular innovation.

b. Core Principles & Technical Advantages
Ultra-Fast Computing Engine: The specially optimized algorithm leverages hardware acceleration to increase docking throughput by orders of magnitude, making screening of billion-molecule libraries feasible.
Quantum Precision Enhancement: The integration of QM-based spectroscopic simulations (including NMR, VCD, and IR) enables highly accurate structural elucidation, resolving critical challenges in drug structure determination and intermolecular interaction characterization. This quantum-level insight significantly improves decision quality across the discovery pipeline, increasing overall drug discovery success rates from ~10% with conventional approaches to over 60%.
Scaffold Novelty Guidance: Molecular design and optimization leverage molecular complexity descriptors derived from matched molecular pair (MMP) analysis, enabling systematic extraction of structure–property relationships. This approach facilitates the discovery of novel chemical scaffolds, allowing exploration beyond the constraints of known active chemotypes and unlocking previously inaccessible regions of chemical space.
Conformational Flip Simulation: A unique algorithm simulates local conformational changes in the protein, exploring more realistic binding pocket flexibility and uncovering potential binding modes missed by rigid docking.
c. Who Is It For & What Problem Does It Solve?
The Challenge: Traditional virtual screening is either too slow (incapable of screening large libraries) or lacks accuracy (high false-positive rate) and heavily relies on known active scaffolds, making it difficult to discover genuinely novel structures.
Our Solution: AlanDockAI™ makes high-precision screening at a "lead-like" level within ultra-large commercial or virtual libraries a reality. It significantly increases the probability of discovering novel scaffold hits, injecting innovation directly into the earliest stages of a project.
d. Typical Workflow
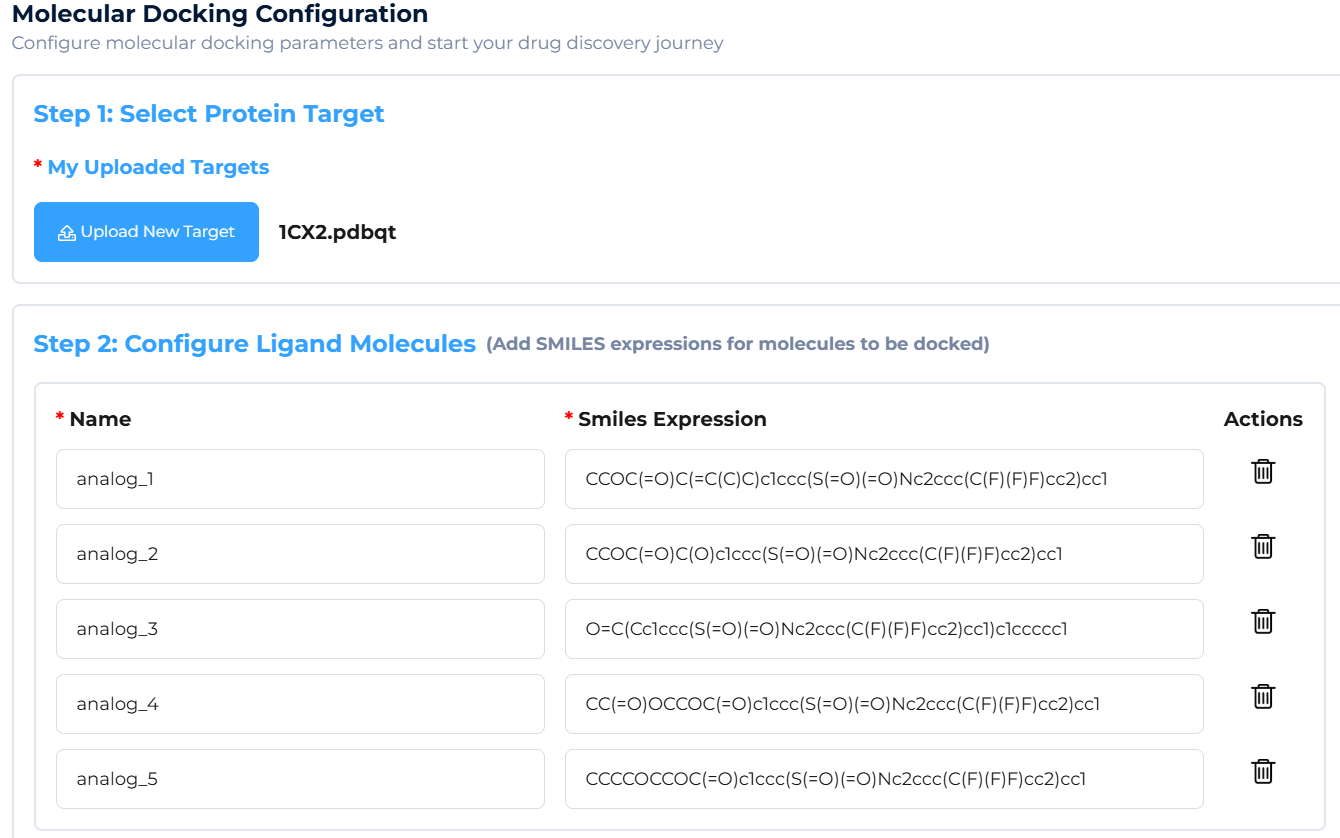
Preparation: Prepare the target protein structure (in PDB/PDBQT format) and the molecular SMILES for screening (single or multiple).
Molecular Generation/Optimization: Generate de novo molecular structures using a well-trained large language model (LLM), followed by targeted molecular optimization guided by ADMET objectives, including potency, selectivity, pharmacokinetics, and safety-related properties.
Output: Obtain a shortlist of meticulously filtered hit compounds with reasonable binding modes and structural diversity, ready for procurement or synthesis and testing.
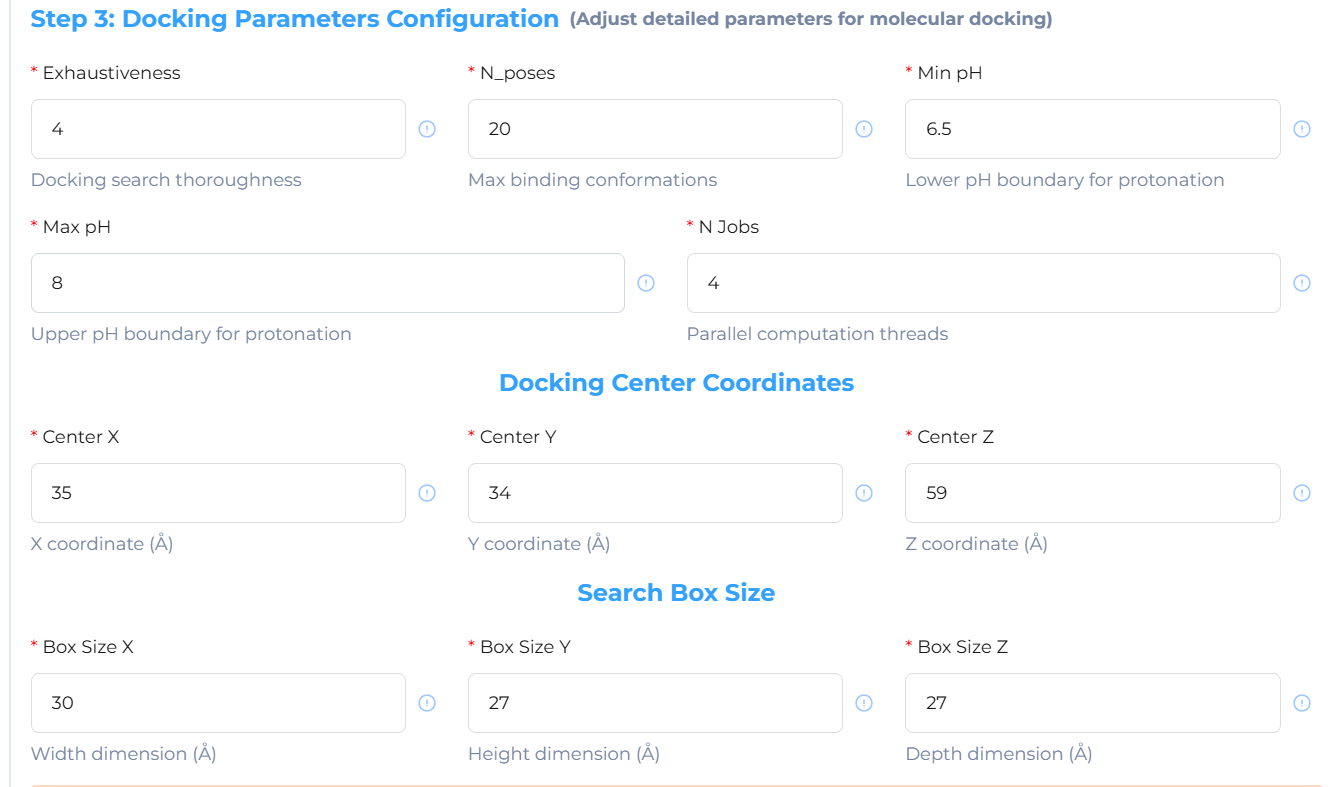
Structure-based Screening: Initiate the docking task; batch docking is completed within minutes to several hours.
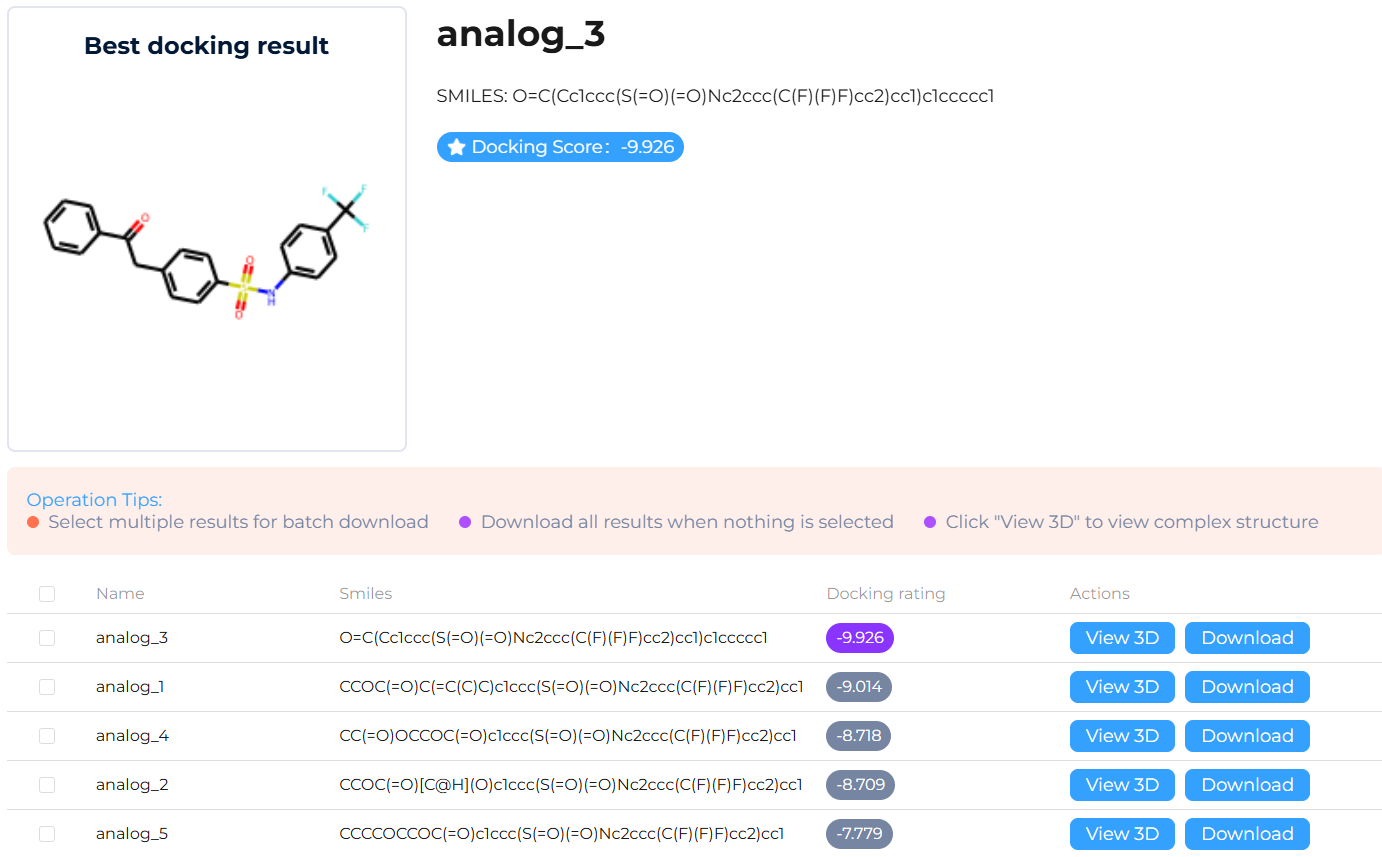
Enrichment: Rank and enrich the results using a comprehensive scoring system (incorporating docking scores) and binding affinity evaluation.
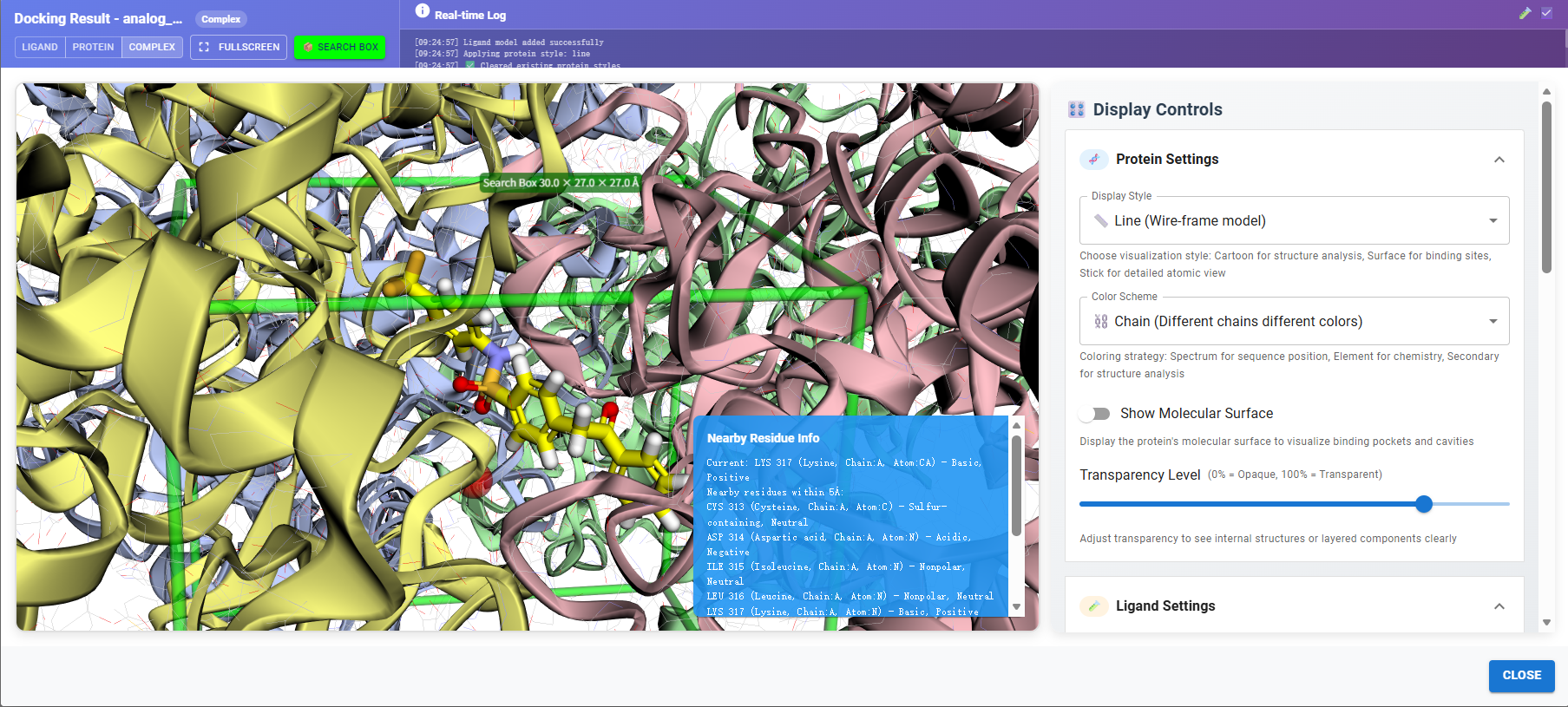
Analysis: Conduct comprehensive visual inspection and analysis of docked binding modes to validate key ligand–target interactions. Generate a binding mode summary that highlights critical interaction features, including hydrogen-bond formation patterns, interaction geometry, and binding consistency across top-ranked compounds.
e. Cost
Each small molecule docking costs 10 credits
f. Showcase
Other AI-Powered Quantum Computing Tool Platforms:
AlanPepAI™: Online Platform for Linear Peptide Sequence Optimization
AlanMolecularAI™: AI-Quantum Computing-Powered Online Optimization Platform for Small Molecules, integrated with proprietary molecular fragmentation technology, enables multi-round ADMET model screening and accelerates the early discovery and optimization of small molecules
To Get More Credits or View Pricing Plans:
Log in and go to My Account > AIPower Platform > Add Credits to Your Account
For Peptide Synthesis Service:
Please click here Peptdie Synthesis